

























Анализ рисков и процессный подход — факторы успеха
Авторы материала:
Елена Зелинская, GMP-эксперт ООО «Ай.Би.Эй»
Александр Белинский, начальник отдела валидации ООО «ЛабПромИнжиниринг»
 Авторы не включили большой теоретической отдел введения, сознавая, что если вы выбрали эту статью, то как минимум, знакомы с понятием, что такое валидация компьютеризированных систем и для чего это нужно.
Авторы не включили большой теоретической отдел введения, сознавая, что если вы выбрали эту статью, то как минимум, знакомы с понятием, что такое валидация компьютеризированных систем и для чего это нужно.
Однако, чтобы говорить на одном языке, хотим напомнить, что входит в определение компьютеризированная система (КС). Взяв только один нормативный документ Решение № 77 Совета Евразийской экономической комиссии «Правила надлежащей производственной практики Евразийского Экономического Союза» от 3 ноября 2016 г. [1], мы встретили там несколько определений КС в разных частях документа:
В соответствии с GAMP 5 [2] компьютеризированная система — это действительно система, которая вбирает в себя все составляющие, которые участвуют в процессе, например:
 Рисунок 1. Схематичное изображение КС
Рисунок 1. Схематичное изображение КС
Несомненным ядром любой КС является программное обеспечение (ПО). Целесообразно четко разделить ПО системное и ПО прикладное. Системное ПО – это операционная система, драйвера, утилиты и т.п. Прикладное ПО – это исполняемые приложения, которые могут быть запущены только с задействованием определенного системного ПО. ИТ-инфраструктура при таком разъяснении перестанет быть загадкой и, проще говоря, может быть определена по формуле «железо + системное ПО».
Категории программного обеспечения КС
Для удобства анализа различных КС в руководстве GAMP 5 предложен подход с выделением различных категорий, которые укрупненно ранжируют такие системы по простоте свой структуры с одной стороны и в части уникальности с другой. Таким образом, представляется возможным сделать фокус на уникальных продуктах, где риск допустить ошибку выше, т.к. и количество пользователей таких продуктов ограничено и, следовательно, возрастает вероятность ненадлежащего функционирования. Например, неработоспособность кнопки «Пуск» в Windows крайне маловероятна, т.к. такой баг вызвал бы многомиллиардный резонанс по числу пользователей во всем мире. Поэтому операционная система (например: Windows) – это самая простая категория, относящаяся к инфраструктурному ПО (категория 1), а вот если вы написали самостоятельный код, обсчитывающий площадь хроматографических пиков или оптическую плотность, то проверят вас 2-3 химика только вашей лаборатории ОКК и при наличии непреднамеренной логической ошибки в расчетах подобный баг способен поставить под сомнения все результаты испытаний с задействованием подобной программы. Это будет категория 5 – ПО, выполненное по индивидуальному заказу. Это полярные категории. Между ними расположено всего две – несконфигурированное ПО (категория 3) и сконфигурированное ПО (категория 4). Категория 2 (т.н. firmware, прошивка) упразднена, т.к. со временем пришло осознание, что прошивка приборов может быть разной сложности.
Категории программного обеспечения в соответствии с GAMP 5:
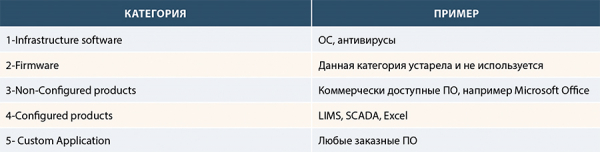 Рисунок 2. Категории программного обеспечения (GAMP 5)
Рисунок 2. Категории программного обеспечения (GAMP 5)
При этом важно понимать, что в реальности для сложных КС, например 1С или ERP SAP вполне могут быть реализованы различные категории одновременно – часть модулей идут в конфигурации по умолчанию (default settings), часть может быть сконфигурировано, а ещё часть – дописано под индивидуальные нужды. Основной фокус валидации, в таком случае, необходимо нацелить на категорию 5 – как наиболее рисковую позицию, которую нужно всесторонне проверить на уязвимости, обработку ошибок, поведение при вводе некорректных данных и т.п.
Анализ рисков или начало начал
Если обратиться к Приложению № 11 (Требования к КС) Решения № 77 Совета ЕЭК «Правила надлежащей производственной практики ЕАЭС» от 3 ноября 2016 г. то первый же пункт этого приложения посвящен управлению рисками:
Управление рисками должно применяться в течение жизненного цикла компьютеризированной системы и учитывать безопасность пациентов, целостность данных и качество продукции. В рамках системы управления рисками решения по объему валидационных испытаний и проведению контролей целостности данных должны основываться на обоснованной и документально оформленной оценке рисков компьютеризированной системы.
Каким образом реализовать данный пункт на практике? Вспомним схему управления рисками из ICH Q9 [3] :
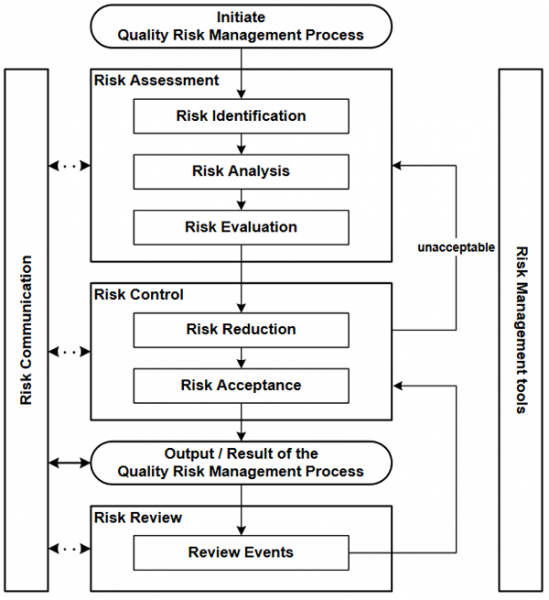 Рисунок 3. Процесс управления рисками качества (ICH Q9)
Рисунок 3. Процесс управления рисками качества (ICH Q9)
В связи с тем, что при валидации КС мы руководствуемся GAMP 5, обратимся к этому документу и посмотрим схему управления рисками:
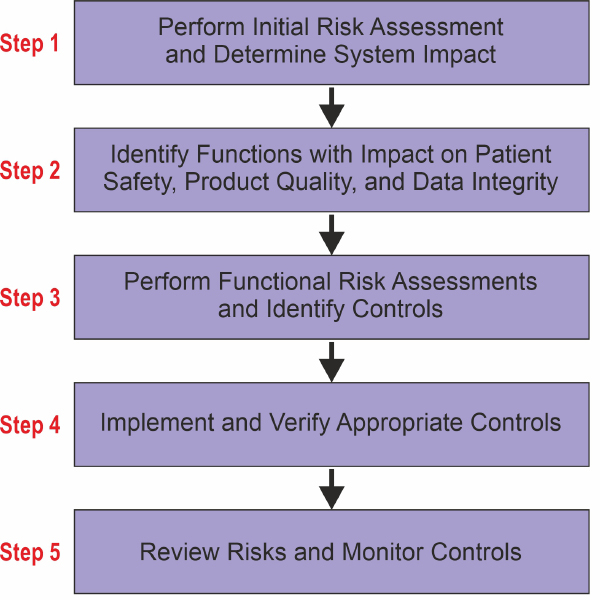 Рисунок 4. Процесс управления рисками качества (GAMP5)
Рисунок 4. Процесс управления рисками качества (GAMP5)
GAMP 5 определяет 5 четких шагов/этапов управления рисками. Теперь попробуем соотнести эти две схемы друг с другом:
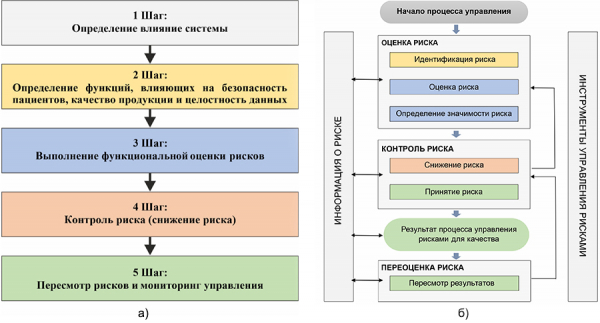 Рисунок 5. Сравнение процессов управления рисками: а) GAMP 5; б) ICH Q9
Рисунок 5. Сравнение процессов управления рисками: а) GAMP 5; б) ICH Q9
Каждый шаг из схемы GAMP 5 окрашен в определенный цвет и в те же цвета окрашены соответствующие этапы из схемы ICH Q9. Мы видим, что соблюдается как общий принцип, так и последовательность и логика в этих двух схемах. Теперь попытаемся разобрать каждый шаг поэтапно и понять какие конкретные шаги необходимо выполнить при прохождения каждого этапа.
Шаг№ 1: Определение влияние системы
На этом этапе необходимо понять и описать бизнес-процессы которые будут реализовываться с помощью КС. (см. пункт «Описание бизнес-процессов»)
Одним из важных моментов этого этапа – это определение попадает ли КС под требования GxP систем. Один из вариантов определения GxP релевантности является заполнение опросника/анкеты. Часто подобные анкеты нацелены только на требования GMP. Да, это наиболее часто встречающиеся требования, но для каждой компании необходимо определить те актуальные требования, которые необходимы именно для вас. Например, GMP/GDP, или GLP/GCP, или GVP. Предлагаем один из вариантов такого опросника:
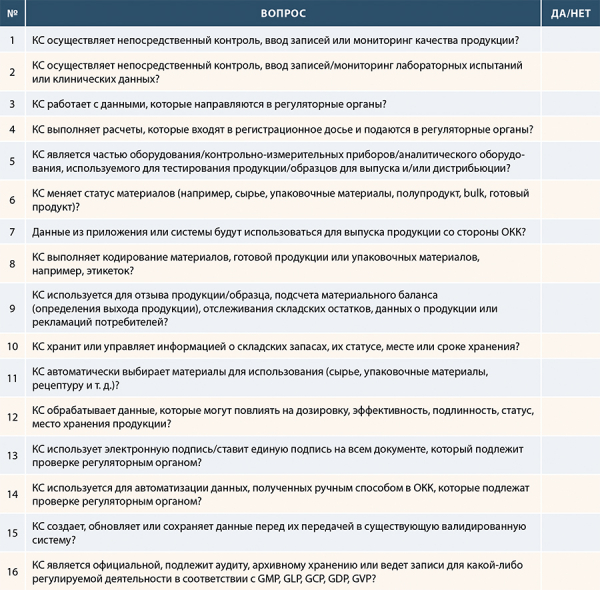 Рисунок 6 Анкета определения GxP релевантности
Рисунок 6 Анкета определения GxP релевантности
Если вы отвечаете «да» хотя бы на один вопрос анкеты, это обозначает, что КС является GxP-релевантной, т.е. попадающей под действия требований GxP и подлежит валидации.
Шаг№ 2: Определение функций, влияющих на безопасность пациентов, качество продукции и целостность данных
Этот этап подразумевает разработку спецификаций (URS, FS etc.). Количество и виды спецификаций зависит от вида КС, которые мы рассмотрели выше. Приведем пример, какие виды СП предлагает GAMP 5 для 4-й категории КС:
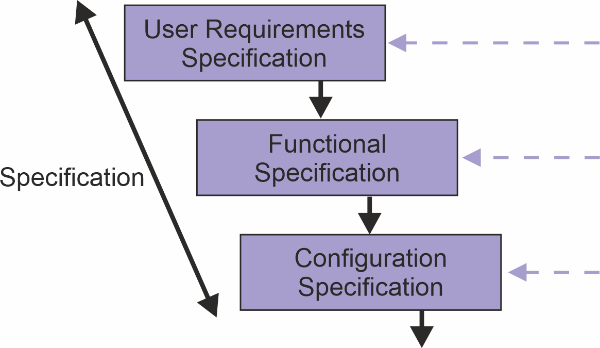 Рисунок 7. Спецификационная часть V-модели для 4-й категории компьютеризированной системы согласно GAMP 5 (источник: ISPE)
Рисунок 7. Спецификационная часть V-модели для 4-й категории компьютеризированной системы согласно GAMP 5 (источник: ISPE)
Хотим обратить внимание на User requirements specification (URS). Если мы вернемся к Приложению № 11 Решения № 77 Совета ЕЭК «Правила надлежащей производственной практики ЕАЭС» от 3 ноября 2016 г., то нам написано:
4.4. URS должны описывать необходимые функции компьютеризированной системы на основе документально оформленной оценки рисков и влияния с точки зрения соблюдения Правил.
Так что первично, URS или оценка риска? Сейчас мы с вами в самом начале управления рисками, где необходимо разработать URS, а Правила GMP нам говорят, что мы должны это сделать уже на основе документально оформленной оценки риска. Из нашего опыта, оценить риск можно только тогда, когда набрана информация об этом риске. Каким же образом можно совместить эти противоположные требования? Мы считаем, что URS – это живой документ, который может актуализироваться в зависимости от вашего продвижения в познании процесса КС. Таким образом, первую версию URS (иногда можно встретить термин – preURS) разрабатывают на основе первичной информации о КС, которую вы бы хотели внедрить, далее определяются риски в соответствии с этим вариантом URS, они оцениваются и URS актуализируется. Таким образом, финальная версия URS основывается на документально оформленной оценке рисков.
Каким образом можно разработать первоначальную URS?
Мы советуем опираться на бизнес-процессы (см. пункт «Описание бизнес-процессов») и проанализировать нормативные документы:
 Рисунок 8. Пример анализа НД для URS
Рисунок 8. Пример анализа НД для URS
К разработке URS необходимо привлекать IT-специалистов, разработчиков КС, других узких специалистов, которые смогут детализировать URS, и по мере необходимости FS (функциональную спецификацию), СS (конфигурационную спецификацию) и представить актуализированное описание системы в соответствии с требованием п. 4.3 Приложения № 11 Решения № 77 Совета ЕЭК «Правила надлежащей производственной практики ЕАЭС» от 3 ноября 2016 г.: Для критических компьютеризированных систем должны быть в наличии подробное текущее описание физических и логических взаимосвязей, потоков данных и интерфейсов с другими системами или процессами, требуемые ресурсы всего компьютерного оборудования и программного обеспечения, доступные меры безопасности.
При разработке URS необходимо решить вопрос будет ли подключение интернета к вашей системе и каким образом вы поступите, например, с обновлениями операционной системы, приложений MS Office, предусмотреть аудиторский след (Auditor Trail) и т.п.
Описание бизнес-процессов
Вместе с тем, когда перед вами откроется реальное ПО, например, 1С – возникнут проблемы. Многие предприятия уже имеют эту систему по линии финансово-экономических отделов и зачастую имеют и штатных ИТ-специалистов, её сопровождающих и, по мере необходимости, дорабатывающих. 1С достаточно хорошо масштабируется и на другие бизнес-процессы предприятия, включая GxP-релевантные. Самое распространённое применение – это управление статусами сырья, материалов и готовой продукции.
Столкнувшись с реальной системой 1С вы с очень большой вероятностью впадёте… в ступор. Схожие эмоции вы испытаете, если попробуете подойти к ERP (SAP, QAD Enterprise, Галактика) или LIMS даже если вы прекрасно знакомы с порядком работы в системе – вот перед вами экран:
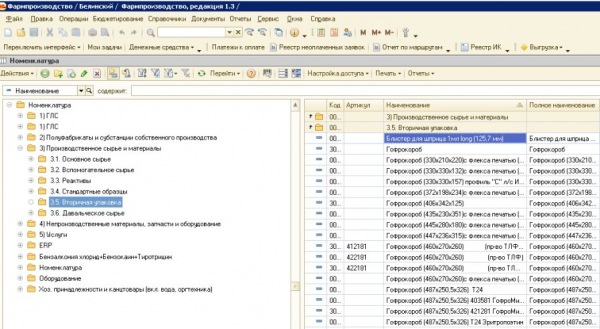 Рисунок 9. Пример пользовательского интерфейса системы 1С (Фармпроизводство)
Рисунок 9. Пример пользовательского интерфейса системы 1С (Фармпроизводство)
Очень быстро выяснится, что, к примеру, выбрав в указанном примере в Справочниках => Номенклатуре => Производственное сырье и материалы => Вторичная упаковка вы упретесь во что-то типа: «Блистер для шприца не может быть выбран». На самом деле как минимум должна присутствовать ролевая модель и статусная модель. И если вы увлечётесь заполнением шаблонных таблиц из GAMP 5, то можете этот весьма существенный момент попросту упустить из виду. Хотя, намёки на это и в GAMP 5, да и в основном тексте GMP/GDP присутствуют. В GMP в Приложении 11 в п.2: «…Весь персонал должен иметь соответствующую квалификацию, уровень доступа и нести определенную ответственность для выполнения возложенных на него обязанностей» (выдел. авторами). В GDP есть п. 47: «Ввод данных в компьютеризированную систему или их изменение должны осуществляться только работниками, ответственными за данный вид работы». Если эти фразы воспринять, как дежурные, то вполне можно пропустить основную суть этих требований. Отчасти намек на разгадку содержится в современных руководствах по целостности данных (MHRA [4] и ГИЛС и НП [5]) – это т.н. DIRA = data integrity risk assessment, оценка риска целостности данных. В обоих документах указано, что процессы, генерирующие данные должны картироваться (mapped out) и отмечается тот, факт, что оценка рисков должна быть сосредоточена на бизнес-процессе [4][5]. Однако, к сожалению, не детализируются, а это по мнению авторов крайне важно, иначе вся ваша работа будет иметь формальный характер и вы можете упустить основные риски и неграмотно провести валидацию.
Существует большое количество методологий моделирования бизнес-процессов, они именуются нотациями. IDEF0, BPMN 2.0. Краткое визуальное представление об этом в контексте фарма можно получить по ссылке.
Приведем пример на стандартных GxP-операций. На любом предприятии есть приемка сырья и материалов. Есть определенный порядок шагов, определенные логические ветвления – взгляните на схему ниже:
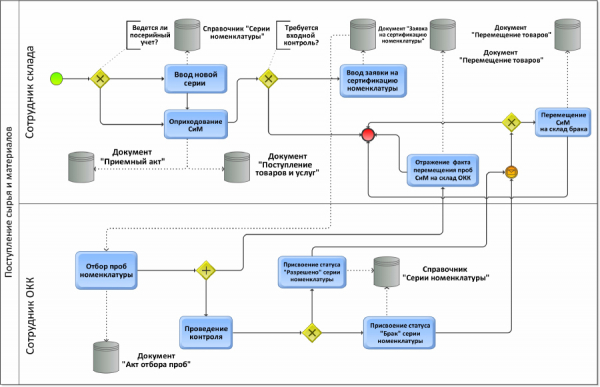 Рисунок 10. Пример моделирования бизнес-процесса в нотации BPMN 2.0 (Поступление сырья и материалов)
Рисунок 10. Пример моделирования бизнес-процесса в нотации BPMN 2.0 (Поступление сырья и материалов)
Даже человеку незнакомому с BPMN 2.0 будут знакомы общие для многих этапы – оприходование сырья и материалов, отбор проб, присвоение статуса «Карантин», «Разрешено», либо статуса «Брак» и т.п. Подразумевается, что при прохождении данного квеcта вы и должны определить рисковые позиции, оценить, что может пойти не так и реализовать это в URS. Если этого не сделать на этапе URS, то разработчик может упустить из виду существенные для вас моменты и «ловить» вы их будете при разработке или уже в продуктивной среде.
Пример другой нотации: IDEF0 – содержит всего два типа элементов – функциональные блоки (прямоугольники) и интерфейсные дуги (стрелочки):
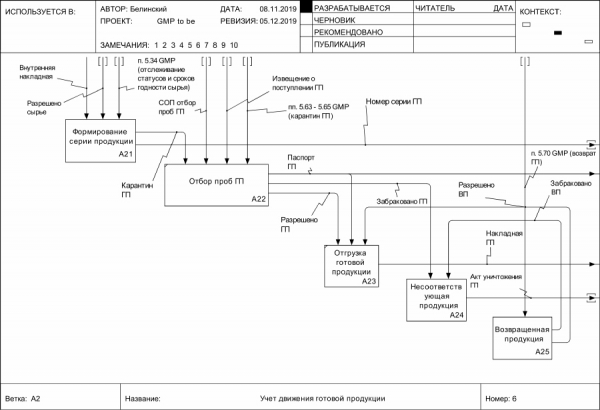 Рисунок 11. Пример моделирования бизнес-процесса в нотации IDEF0 (Учет движения готовой продукции)
Рисунок 11. Пример моделирования бизнес-процесса в нотации IDEF0 (Учет движения готовой продукции)
На рисунке выше изображен процесс учета движения готовой продукции. Сверху – стрелки управления – что запускает тот или иной вид деятельности и на предмет соответствия каким критериям он осуществляется. Например, при формировании серии продукции основанием в примере служит внутренняя накладная (на разных предприятиях наименования могут варьировать), а также статус сырья «Разрешено». В любом другом статусе эта функция должна быть заблокирована. Кроме того, можно увидеть и наличие регуляторного пункта (п. 5.34 GMP, обязывающий отслеживать статусы и сроки годности сырья и материалов), иначе можно использовать сырье разрешенное, но… с истекшим сроком годности. Выходом (стрелочками справа) данной функции является сформированная серия готовой продукции и её статус «Карантин», для того чтобы могла быть доступной следующая функция – отбор проб готовой продукции.
Без визуального представления очень сложно в повествовательном режиме описать такие бизнес-процессы.
Вышеприведенные нотации – это средства моделирования, которые обеспечены программными продуктами, автоматизирующими этот процесс. Так, благодаря системам моделирования (в указанных примерах для нотации BPMN использовалась свободно распространяемая BPMS ARIS Express, а для IDEF0 – аналогично, свободно распространяемый программный продукт Ramus) можно провести оценку как количества функций, так и количества объектов системы (формируемых документов) и сделать это целесообразно до этапа разработки КС.
И вот именно прохождение таких (или подобных схем) сгенерирует вам невымышленные риски, которые можно «упаковать» в таблицы, рекомендуемые GAMP 5 (FME(C)A).
Без подобного моделирования бизнес-процессов вы в принципе не сможете сформировать внятное задание для разработчика и можете погрязнуть в бесконечных процессах внедрения и тестирования, часто заканчивающихся возвратом практически на исходную позицию. Ведь программный продукт не будет работать «по щучьему велению» и при «сочинении» программы произвольным способом вы сформируете большие риски, выражающиеся, например, в том, что некоторые акты в принципе не сможете сформировать, а ряд статусов никогда не будет присвоен. Что происходит на практике, когда отсутствуют URS, FS и какая-либо формализация бизнес-процессов. Даже если кто-то из фланга качества попытается «натянуть сову на глобус» – это будет только «фиговый листочек для инспектора» – никакой практической пользы без глубокой проработки эта деятельность иметь не будет.
Продолжаем разговор про URS и FS
На практике зачастую возникает необходимость для уже работающих систем ретроспективно воссоздать URS и FS. Если система давно находится в эксплуатации (Legacy System- в терминологии PIC/S [6]) может быть несколько достаточно простых и, вместе с тем, реализующих регуляторные ожидания подходов:
- в случае если поставщик/разработчик системы был внешним, п. 4.4. Приложения 11 GMP вы вполне можете выполнить, призвав на помощь бухгалтеров. Они не могли закрыть работы без договоров и актов выполненных работ. Поднимите договора, посмотрите их условия, где указан объем внедрения. И представьте это в виде ретроспективного URS. Вы именно за это в своё время заплатили деньги.
- в случае, если разработчик ПО внутренний и договора с ним нет, придётся URS разработать ретроспективно и тут тоже предлагаем два пути. Когда вы закончите работу с FS в соответствии с принципами, изложенными ниже – вы получите детализированный документ (схемы GxP-релевантных бизнес-процессов и описаний к ним). Можно назвать его URS/FS, как будто бы вы именно это и задумывали с самого начала. Другой вариант, всё же сделать отдельный документ, например, описав базово эти же функции.
А вот текущее описание FS мы рекомендуем выполнить со всей тщательностью. Потому что в большинстве случаев владельцы таких систем, уже находящихся в использовании, даже не подозревают, чем они владеют. С момента первичного внедрения уже могла поменяться операционная система, могли быть выполнены доработки функционала по отношению к первичному объему внедрения, поэтому обойтись описаниями в договорах n-летней давности тут не получится.
FS должна быть разработана в тесной кооперации с ключевыми пользователями системы и, по возможности, с её разработчиками. Для того формализованные бизнес-процессы следует описать с применением инструментария, приведенного на рис. 12:
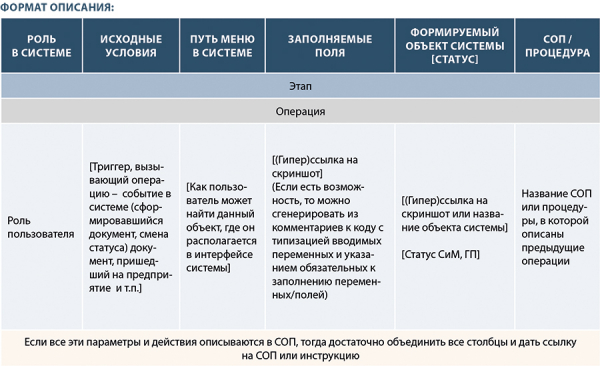 Рисунок 12. Пример шапки таблицы FS
Рисунок 12. Пример шапки таблицы FS
Используя данную таблицу, вы последовательно можете описать все функции, ранее смоделированные в нотациях IDED0, BPMN 2.0 или просто в виде простой алгоритмической схемы. Для действующей системы каждый «прямоугольничек» (функция) по схемам должен быть описан с указанием роли пользователя (кто конкретно выполняет ту или иную функцию, под какой учетной записью она доступна), какие исходные условия должны наступить, чтобы эта функция была осуществима, какой пусть в меню системы, какие заполняемые формы и/или поля, какие объекты системы формируются и какие статусы присваиваются. Такой подход в целом применим практически к любой системе.
И вот только тогда, когда опишите подобным образом все ваши функции, только с этого момента можно приступить к функциональному анализу рисков. Даже совпадение терминов получилось. О каком функциональном анализе рисков можно говорить до того момента, пока не знаем не то, что функциональный состав системы, но даже общее количество функций.
Конечно любые последующие изменения в системе следует оформлять именно в виде URS, (со слов представителей бизнеса), затем на их основании создавать описание бизнес-процессов и FS по примеру выше, т.е. строить дом с фундамента, а не с крыши. Тогда вы и требование в отношении контроля изменений выполните, не потеряв при этом управляемость в системе. При этом любые изменения следует «обкатать» сначала в тестовой среде. Системные администраторы организуют вам для этого отдельный сегмент сети (т.н. VLAN).
В отношении тестовой среды есть намек в GMP EU, но не вполне точный перевод на русский язык нивелировал его. Сравните оригинальный текст: “4.7 Evidence of appropriate test methods and test scenarios should be demonstrated. Particularly, system (process) parameter limits, data limits and error handling should be considered. Automated testing tools and test environments should have documented assessments for their adequacy”
А вот перевод этого пункта согласно [1]: «4.7. Следует представить доказательства соответствия методов и схем тестирования компьютеризированной системы. В частности, должны быть рассмотрены пределы параметров системы (процесса), границы данных и обработка ошибок. Следует документально оформить 5 оценку соответствия применения автоматизированных средств тестирования и режимов их работы».
Тестовая среда или, на языке ИТ-специалистов «песочница» (sandbox) – вполне конкретное понятие. Но на этот термин непросто выйти исходя из «режима работы средств тестирования». Поэтому, конечно, полезно изучать документы в сопряжении с оригиналом. Правда есть некоторые моменты, которые и в оригинале не являются прозрачными. Поэтому предложим небольшое подспорье, которое неожиданно пришло от GLP – это руководство OECD GLP 2016-го года [9], которое изложено практически по пунктам Приложения 11 GMP, но содержит несколько развернутых комментариев. При участии одного из авторов выполнен перевод указанного руководства на русский язык. Руководство предоставит значительно более глубокое понимание подходов при разработке и валидации КС, нежели чем Приложение 11 GMP.
Шаг №3: Выполнение функциональной оценки рисков
По схеме ICH Q9 этот этап называется оценка риска. Это этап, где происходит ранжирование рисков и именно его обычно представляем при упоминании словосочетания оценка риска. Этот этап не оторван от предыдущих двух, а является их логическим продолжением.
Берутся по очереди пункты из FS и по каждому пункту определяются риски, (опасность) и их последствия, какие проблемы могут возникнуть для процесса, если этот пункт не будет реализован по той или иной причине. Таким образом образуется список рисков. Эти риски необходимо про ранжировать:
- Высокая серьезность (угроза жизни пациента)
- Средняя серьезность (возможен вред здоровью пациента)
- Низкая серьезность (отсутствует влияние на здоровье пациента)
- Высокая (чаще чем 1 раз в месяц)
- Средняя (чаще чем 1 раз в 6 месяцев)
- Низкая (реже чем 1 раз в 6 месяцев)
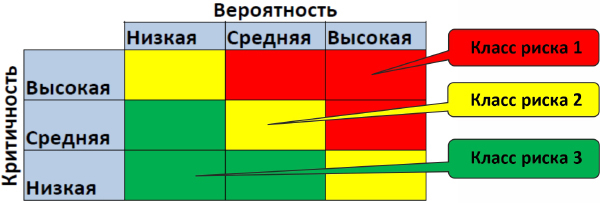 Рисунок 13. Определение класса риска (GAMP5)
Рисунок 13. Определение класса риска (GAMP5)
4. Определяется Обнаруживаемость (вероятность того, что ошибка будет замечена до наступления вреда)
- Низкая-не обнаружено системами компании
- Средняя-один механизм для обнаружения
- Высокая- более одного механизма для обнаружения
5. Определяется Приоритетность риска (оценка первичного риска) по алгоритму: Класс риска умножить на Обнаруживаемость
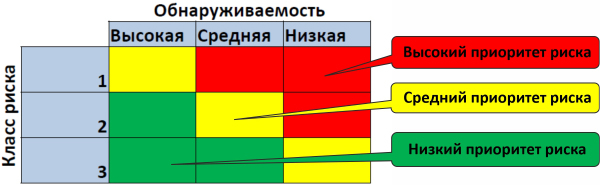 Рисунок 14. Определение приоритета риска (GAMP5)
Рисунок 14. Определение приоритета риска (GAMP5)
Таким образом, можно заполнить следующую таблицу:
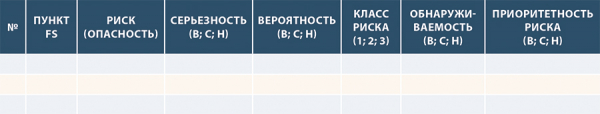 Рисунок 15. Пример шаблона таблицы оценки рисков
Рисунок 15. Пример шаблона таблицы оценки рисков
Примеры частично заполненного шаблона анализа рисков на основании формализованных функций в FS:
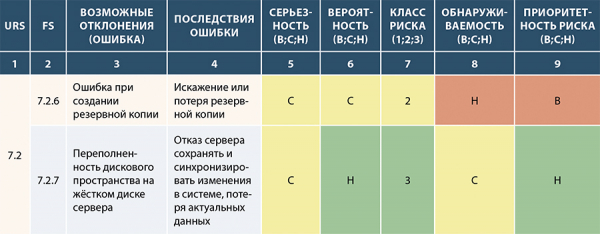 Рисунок 16. Пример №1 частично заполненного шаблона анализа рисков
Рисунок 16. Пример №1 частично заполненного шаблона анализа рисков
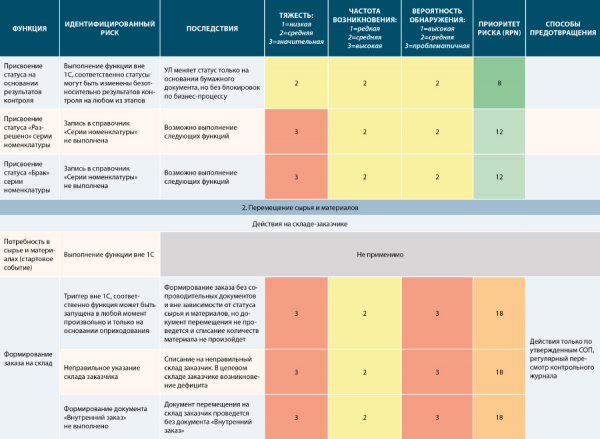 Рисунок 17. Пример №2 частично заполненного шаблона анализа рисков на основании формализованных функций в FS (согласно шаблону на рис. 12)
Рисунок 17. Пример №2 частично заполненного шаблона анализа рисков на основании формализованных функций в FS (согласно шаблону на рис. 12)
Шаг№ 4: Внедрение и проверка соответствующих элементов управления
На этом этапе идет разработка мероприятий по снижению неприемлемых рисков, выявленных на этапе ранее, и контроль того, были ли принятые меры эффективными в достижении требуемого снижения риска.
На этом же этапе можно определить какие виды тестирования вы будете выполнять.
Пример заполнения таблицы:
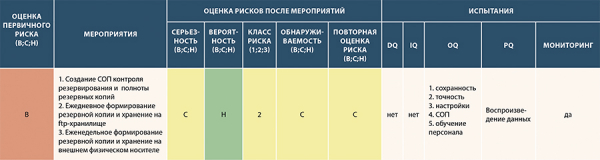 Рисунок 18. Пример частично заполненного шаблона анализа рисков по определению мероприятий, снижающих высокий риск
Рисунок 18. Пример частично заполненного шаблона анализа рисков по определению мероприятий, снижающих высокий риск
Тестирование системы
На данном этапе необходимо разработать протоколы. В связи с тем, что на ранних этапах вы четко определились со всеми видами тестов, разработка протоколов не будет сложной.
Мнения авторов разделились при обсуждении вопроса выделяются ли стации IQ, OQ, PQ при валидации КС. Одному привычней позиция выделения этих стадий, а с альтернативной позицией по этому поводу вы сможете ознакомиться по ссылке [7]. Однако, это не меняет общих подходов к валидации КС и понимания необходимости тестирования в целом. Мы сочли, что данная ситуация будет интересной для читателя и он может самостоятельно выбрать вариант, который ему ближе.
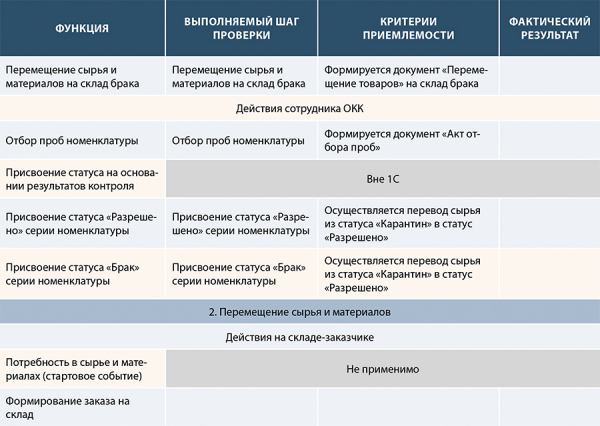 Рисунок 19. Пример частично заполненного шаблона ключевых тестовых сценариев на основании формализованных функций в FS и проведенного анализа рисков (на основании анализа рисков, см. рис. 17)
Рисунок 19. Пример частично заполненного шаблона ключевых тестовых сценариев на основании формализованных функций в FS и проведенного анализа рисков (на основании анализа рисков, см. рис. 17)
Как вариант, вы можете определить основные группы тестирования в зависимости от категории ПО КС (пункт «Шаг№ 1: Определение влияние системы»):
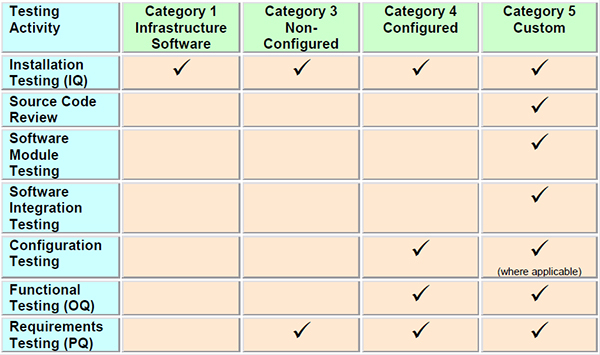 Рисунок 20. Пример распределения тестирования при валидации КС в зависимости от категории
Рисунок 20. Пример распределения тестирования при валидации КС в зависимости от категории
На этом же этапе необходимо разработать СОП. Если вы отнеслись ответственно к предыдущим этапам у вас уже сформировался перечень необходимой документации.
Нормативный документ, который регламентирует перечень СОП для КС это GCP ICH E6(R2) [8] пункт 5.5.3(b):
- установка и настройка системы;
- тестирование рабочих характеристик (параметров) системы;
- сбор данных и работа с ними;
- обслуживание системы;
- защита системы и контроль изменений;
- резервное копирование данных;
- восстановление данных;
- планирование аварийных ситуаций;
- вывод системы из эксплуатации.
Какие в принципе должны быть процедуры в отношении информационных активов хорошо излагают ИТ-стандарты – ISO 20 000 (ITSM = IT Servie management – управление ИТ-услугами) и ISO 27 000 (ISMS – Information Security Management Systems или СУИБ = системы управления информационной безопасностью). СОПы по использованию системы – это описание бизнес-процессов. По сути, надлежащим образом описанная FS (см. рис. 12) формирует вам готовый СОП.
После выполнения всех тестов, оформляется отчет по валидации который как минимум:
- рассматривает процесс всех этапов валидации;
- подтверждается выполнение плана;
- подводятся итоги;
- подтверждается контроль отклонений;
- подтверждается пригодность КС для использования по назначению;
- обосновывается передача данных для использования в среде GxP.
Шаг № 5: Пересмотр рисков и мониторинг управления
Во время периодического обзора систем необходимо подтвердить, что приоритетность риска все еще актуальна и что меры контроля все еще эффективны. Частота пересмотра рисков должна основываться на уровне риска.
Заключение
В итоге, как вариант у вас может сформироваться следующая форма, где каждый цвет столбца соответствует определенному этапу процесса валидации:
 Рисунок 21. Пример общей заполняемой формы по валидации КС
Рисунок 21. Пример общей заполняемой формы по валидации КС
Часто для подобной формы применяется термин TM (Traceability matrix, матрица прослеживаемости требований). Это конечно не единственный вариант реализации подобной матрицы, но суть подобного инструмента всегда неизменна – проследить, что по мере разработки и внедрения системы «не уронили» то или иное требование, «не забыли» тот или иной риск. И если где-то на ранних этапах (URS, FS) вы обозначили рисковую позицию, то на более поздних этапах тестирования нужно понимать, где именно эта позиция протестирована, какое именно внимание мы ей уделили в ходе всего жизненного цикла КС.
В соответствии с пунктом 4.3 (11 Приложение 77 Решение ЕЭК) [1] должен быть реестр всех КС с указанием их функциональности. А для критических КС должны быть описание взаимосвязей, потоков данных и интерфейсов с другими системами или процессами, ПО.
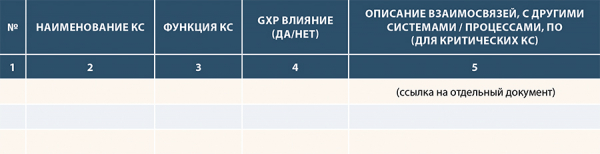 Рисунок 22. Пример формы реестра КС
Рисунок 22. Пример формы реестра КС
Напоминаем этапы Валидации КС:
- разработка перечня всех компьютерных систем;
- определение попадает ли КС под требования GхP;
- описание бизнес- процессов;
- разработка спецификаций;
- разработка перечня рисков;
- функциональная оценка рисков, при необходимости разработка мероприятий по снижению рисков;
- на основе анализа рисков разработка перечня и методик тестов;
- разработка протоколов валидации, разработка СОП, обучение персонала;
- проведение тестирования КС;
- пересмотр рисков и мониторинг управления.
Вот такие базовые практические подходы могут быть предложены исходя, с одной стороны, из современных регуляторных требований и ожиданий, а, с другой стороны, исходя из нашего практического опыта.
Список литературы
[1] Решение № 77 Совета Евразийской экономической комиссии «Правила надлежащей производственной практики Евразийского Экономического Союза» от 3 ноября 2016 г.
[2] ISPE GAMP 5: A Risk-Based Approach to Compliant GxP Computerized Systems
[3] ICH guideline Q9 on quality risk management, September 2015EMA/CHMP/ICH/24235/2006 Committee for Human Medicinal Products
[4] MHRA ‘GXP’ Data Integrity Guidance and Definitions, March 2018
[5] ГИЛС и НП, Целостность данных и валидация компьютеризированных систем
[6] PIC/S Guidance: GOOD PRACTICES FOR COMPUTERISED SYSTEMS IN REGULATED “GXP” ENVIRONMENTS
[7] https://pharm-community.com/2020/10836/
[8] E6(R2) Good Clinical Practice: Integrated Addendum to ICH E6(R1) Guidance for Industry U.S. Department of Health and Human Services Food and Drug Administration ; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER) March 2018
[9] ENV/JM/MONO(2016)13 Application of GLP Principles to Computerised Systems





